If you are confused by the many acronyms related to regulatory submissions, you are not alone. A variety of data from disparate functions must come together to provide a common understanding of a product’s anticipated safety and efficacy. A cohesive submission plan incorporating the GCP, GDP, GLP, GMP, and GVP perspectives helps ensure a quality submission and increase the likelihood of approval.
A quality regulatory submission requires timely input and an appropriate level of detailed GxP data—a significant volume of data for later-stage submissions—from scientists across multiple functions of an organization. The regulatory submission lead defines the strategic global regulatory vision and leverages input from submission team members representing manufacturing, non-clinical, and clinical to compile a regulatory submission. Each function contributes key data that adheres to GxP quality standards specifically defined for working in a regulated environment.
Working seamlessly together across many functions to gather and properly position GxP data to ensure consistent messaging is integral to producing a high-quality, coherent, and concise submission that increases the likelihood of approval in one or more countries. What follows is a primer to turn the alphabet soup of GxPs into a cohesive submission plan and a checklist to help ensure a quality submission. Scientists who are new to regulatory submissions will find it helpful to better understand how their data fits into the overall submission. An experienced regulatory professional can share this primer with colleagues on their regulatory submission team.
Quality First
GxP is an acronym for Good “x” Practice, where “x” can represent many different quality standards; for example, Good Clinical Practice, Good Laboratory Practice, or Good Manufacturing Practice. The purpose of adhering to GxP is to show how quality has been established, monitored, and documented throughout the multiyear product development process. A company culture with a quality mindset positively influences all aspects of the business starting with the Quality Manual and extending into manufacturing as well as regulatory submissions.
Functional teams must use high scientific and ethical standards to collect the data that goes into a regulatory submission. A quality submission is more likely to gain approval. An organization that receives a Clinical Hold for an early stage submission or a Refusal to File for a late stage submission has not done enough due diligence in formulating the submission document. This can result in rework and additional work, time, and expenses to resubmit. The delay in getting to market could also adversely affect the company’s reputation.
Deciphering the Alphabet Soup of GXP Acronyms
Harmonization of a common format for electronic submissions worldwide, defined as the Common Technical Document (CTD), has minimized the need to customize the information for submissions to regulatory authorities.
The three most commonly referenced acronyms that represent the quality requirements for data included in a regulatory submission are GCP, GLP, and GMP. The U.S. FDA, ISO, ICH, and the EU Regulations and Directives have established guidelines to define these terms and how they should be applied to drug development.
GXPS Mapped to the CTD Triangle
GxPs play a key role in providing quality data that goes into the submission. The complex interdependencies across functional areas must be clearly defined and managed for the submission to adequately support product labeling and approval. Regulatory agencies worldwide may require or strongly recommend using the CTD format for regulatory submissions. Organizational functions―grouped by quality, non-clinical, and clinical―contribute to the ICH CTD as outlined in the CTD Triangle shown in Figure 1. Figure 1 also illustrates how GxP roles are mapped to the CTD Triangle.

Module 1 (M1) generally contains regional-specific administrative information managed by Regulatory. M1 content includes forms, cover letters, certifications, patent information, labeling, references, meeting correspondence, application status, etc. Select GMP, GLP, and GCP data is contained in some of the M1 components, including the Investigator’s Brochure and product labeling.
Module 2 (M2) requires summaries of the GMP, GLP, and GCP data from M3, M4, and M5, respectively. Key documents in M1 and M2 are often the last elements of the submission to be completed since they depend on input from other modules.
Module 3 (M3) includes GMP data derived from the Chemistry, Manufacturing and Controls (CMC), or Technical Operations function; for instance, stability, analytical validation, impurities, pharmacopoeias, and specifications related to drug substance and drug product. GMP defines the quality control and quality assurance standards for manufacturing facilities and processes that produce goods including drug substances and drug products.
GLP data derived from preclinical and/or non-clinical studies are presented in Module 4 (M4). Example study data may include carcinogenicity, genotoxicity, toxicokinetics, pharmacokinetics, toxicity, reproductive toxicology, biotechnology, pharmacology, immunotoxicology, etc. GLP provides guidance for conducting preclinical and nonclinical research with robust protocols and coherent record keeping, producing quality laboratory data.
GCP data derived from clinical studies, such as clinical safety and efficacy data and pharmacovigilance information, are presented in Module 5 (M5). GCP guides the ethical and scientific quality standards to ensure the safety of participants and quality data collection during clinical trials.
GVP focuses on the collection of data related to detecting, assessing, and preventing adverse events to increase drug safety. GVP primarily resides in the risk management plan (RMP), which maps to summary data in M2, quality data in M3, non-clinical data in M4, and clinical data in M5. (This site offers in-depth GVP guidelines from the EMA.)
All documentation provided in a submission containing manufacturing, laboratory, and clinical data should adhere to Good Documentation Practice (GDP). GDP guides the development, review, approval, and retention processes of regulatory controlled documents including submission documents. While ICH does not define GDP, best practice standards describe the processes for creating, reviewing, modifying, approving, and maintaining documents. (This site holds thorough GDP guidelines.)
Roles Within the Submission Lifecycle
The submission development plan must be aligned across the many functions that contribute GxP data. A clear understanding of the responsibilities of each contributing function and the interdependencies between key documents and data across modules will help determine which sections of the submission can be completed early and which sections are likely to become available just in time for the submission. These interdependencies can vary based on the stage of product development (early versus late stage) and by the specific product development challenges. For example, in some cases, CMC product stability for an early stage submission is on the critical path and must be complete before other work can begin. However, for late stage submissions, clinical data from a Phase III clinical trial is often on the critical path.
Figure 2 depicts how each GxP function plays a key role at different stages of a regulatory submission. Successfully managing the delicate balance of interdependencies among functional components of the CTD will help produce a timely, more robust submission document.
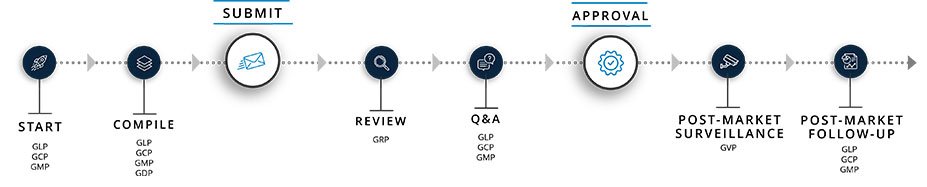
Characteristics of a High-Quality Submission
New companies working on their first submission and individual team members new to submissions often struggle with the complexity of bringing the many disparate parts of the CTD together into a coherent whole. Mature organizations that have completed multiple submissions may want to further optimize their approach to producing high-quality submission documents. Even organizations with extensive regulatory experience can struggle with the complexity of creating strategies, integrating project schedules, and managing stakeholders. A dedicated project leader, orchestrating these multiple activities and resources, can optimize and drive the complicated process.
Quality Submission Checklist
The following checklist serves as a guide to integrate the various GxP functions contributing to the submission to ensure a higher quality submission document and to minimize questions from reviewers. An experienced regulatory professional can use this list as a starting point and work with the submission project manager to help the submission process go as smoothly as possible.
1. A complete and consistent submission document format provides a positive first impression, increasing the likelihood of acceptance.
Lead function: Regulatory Operations
- GDP best practices are followed.
- All dossier sections and supporting documentation are complete.
- Document structure adheres to the CTD.
- Grammar and spelling are correct.
- Formatting adheres to the criteria outlined by the regulatory authority.
- Hyperlinks successfully link to the correct source documentation.
2. The submission document content plan must be clear to regulatory authority reviewers.
Lead function: Regulatory Strategy & Medical Writing
- Target Product Profile (TPP) has been discussed with the regulatory agency to confirm the product development strategy prior to submission.
- Global product development strategy has been defined to inform future submissions.
- Key messaging in the submission document is clear to support product labeling and approval.
- Level of detail for data is sufficient for approval (providing too much data may put the company’s intellectual property at risk in certain countries).
- A well-thought-out plan is in place to rapidly address any potential deficiencies.
3. GxP standards must be defined and adhered to for the submission development process.
Lead function: Regulatory Submissions
- The submission team members are clear on their respective roles and capable of fulfilling their responsibilities for contributing to the submission.
- An acronym list is compiled by the team and used consistently throughout the submission.
- GMP, GLP, and GCP procedures for data collection are well documented and followed.
- The document review and approval process is clearly defined and agreed upon.
Written by: Susan Carino, MBA, MS, PMP, RAC – Principal Consultant
Integrated Project Management Company
Service: Regulatory & Quality
Industry: Life Sciences
"*" indicates required fields